
F1RST UP®: ACCELERATING CLINICAL TRIAL START-UP
Despite the pressing need to bring novel therapies to patients with renal impairments, the development time and cost to bring new drugs and medical devices to market has actually increased over the past decade. Frenova® Renal Research is reversing this trend with F1RST Up®, a more efficient ecosystem of clinical research investigators and leaders in business and medicine. By eliminating traditional clinical trial bottlenecks and implementing a range of patient- focused protocols, Frenova is dramatically accelerating the pace of innovation
The mission of Frenova Renal Research (Frenova) is to advance the science of renal therapies and help bring novel treatments to market for patients with renal impairment. In bringing novel therapies to patients, speed is paramount: speed to approval, speed to product launch, speed to greatest market penetration and adoption. Delays in any step of the approval or product launch pathways result in untreated patients, incremental costs, shortened periods of patent protection, and lost revenue opportunities. Frenova facilitates speed to approval by leveraging F1RST Up™, an exclusive alliance of clinical trial sites dedicated to renal research and designed to accelerate clinical trial start-up and patient enrollment. Through a combination of F1RST Up, Frenova’s relationships with 260 other clinical research site partners, and Frenova’s deep understanding of patients with renal impairment, Frenova also facilitates speed to product launch and commercialization.
In a typical approval pathway, an extensive series of clinical trials to demonstrate safety and efficacy are required before new medicines or medical devices can be introduced into the market. Clinical development is increasingly more expensive and more time consuming. The average clinical development cycle time for a new molecular entity (NME) was 6.5 years during 2000-2013. In 2018, it is closer to 7.5 years. The Tufts Center for the Study of Drug Development (CSDD) published a report in 2014 indicating “the cost of developing a prescription drug that gains market approval at $2.6 billion, a 145 percent increase, correcting for inflation, over the estimate the center made in 2003.”1 EvaluatePharma, an industry leader in consensus forecasting, reported in 2017 that the average cost of developing an NME from discovery to approval is $4 billion.2 Clinical development is the largest expense along the approval pathway. A 2017 report by Nature Reviews Drug Discovery indicated that each additional month in a typical phase III trial added a median expense of $671,000 to developmental costs.3
Several trends are driving increases in clinical development timelines and costs including:
- Increasing protocol complexity
- Complex data collection burdens
- Inefficient site selection
- Challenging site activation processes
The last decade has seen a dramatic increase in protocol design complexity. Research conducted by CSDD characterizes this trend.4 Part of the reason protocols are more complex in 2018 is the need to improve our understanding of chronic diseases and their economic impact, including kidney disease. Gaining better insight into chronic disease requires collecting more clinical data than we have in the past. Consequently, this places greater demands on research staff. CSDD’s research indicates the work burden on research sites increased as much as 73 percent between 2002 to 2012, outpacing growth in the total number of procedures performed per protocol. Understandably, research reveals a high turnover rate among investigators conducting trials, with about 40 percent each year choosing not to conduct any further trials, citing regulatory compliance burden and a challenging operating environment.5
One of the most persistent bottlenecks in clinical research is study start-up. This includes protocol feasibility assessment, site identification and selection, and site activation. CSDD found that the process of preparing a site to enroll patients typically takes one year.6 Site selection is typically a time-consuming, unsophisticated, and inefficient process. The methods used for selecting sites typically “lack verification and are slow, taking 3.2 months, on average.”7 One reason for this is the highly fragmented nature of the traditional clinical research site community. Another reason is the growing investigator community that includes a sizable percentage of first-time researchers. CSDD estimated there were over 35,000 Food and Drug Administration-regulated investigators globally in 2016, with 32 percent being new to clinical research.8
Site activation is another persistent holdup in clinical research and often includes redundant processes and documentation. For each clinical trial, sites typically provide responses to feasibility surveys, copies of medical licenses, training records, curriculum vitaes, etc. Sites must also negotiate confidentiality agreements, clinical trial agreements, and budgets. Contract and budget negotiations are often identified as the major hurdles to starting new studies. Research sites often lack experienced commercial staff who can properly evaluate terms and conditions of trial contracts and budgets, resulting in suboptimal outcomes for the site. In a survey of more than 400 investigative sites, CenterWatch found that contract and budget negotiations are protracted and fraught with delays, contributing to slow trial start-up timelines.9 In the same study, it was revealed that, on average, nearly 11 percent of sites selected are never activated. The primary reasons cited were budgeting and contracting problems.
Patient enrollment accounts for between 35 and 50 percent of a trial’s timeline.10 Even though a site may be selected and activated, patient enrollment performance varies widely from site to site, making enrollment another area of persistent challenge for trial timelines and a driver of added costs. A 2014 study by CSDD concluded that 48 percent of clinical research sites fail to achieve their patient enrollment targets, including 11 percent that fail to enroll a single patient (Figure 1).11 CSDD also found that 53 percent of studies examined had extended timelines, with one in six studies taking more than twice as long as originally planned.12 Other studies have found that 90 percent of all clinical trials fail to meet their original timelines.
FIGURE 1 | Achievement rates for a typical multicenter clinical trial
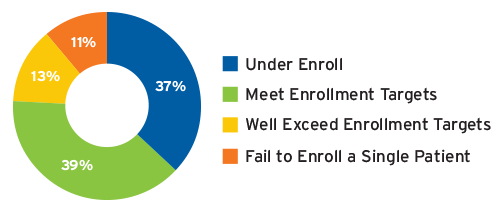
Source: Tufts CSDD, 2014, csdd.tufts.edu
In a 2016 survey of clinical research professionals, CSDD assessed clinical trial start-up practices and performance. The top-scoring study start-up process enhancements were:13
- Central institutional review board (IRB) approval process (94.5 percent)
- Standardized contracting for industry-sponsored clinical trials (90.1 percent)
- Track site’s mandatory terms and previously negotiated language in greater detail to quickly get to study specific negotiations and/or determine if contracting will be a deal breaker (86.3 percent)
Frenova’s answer to addressing process enhancements and tackling other clinical trial inefficiencies typically experienced during a new clinical trial is F1RST Up (Frenova Rapid STart Up).
Launched in early 2016 and expanded in 2017, F1RST Up is an exclusive alliance of 23 clinical trial sites dedicated to renal research. The alliance includes 63 physician investigators and thought-leaders from across the country. F1RST Up investigators and their practice partners provide care to approximately 170,000 chronic kidney disease patients and 15,000 end-stage renal disease patients, collectively. A dedicated team of Frenova’s clinical research professionals manage and help orchestrate F1RST Up activities in close collaboration with alliance partners.
F1RST Up sites were invited to join the alliance following several months of meticulous planning and review of historical trial performance data. Candidate sites were assessed on:
- Experience
- Investigator engagement
- Number of principal investigators and sub-investigators
- Size of practice and patient populations
- Systems and processes
- Business strategy and acumen
- Number, turnover, and experience of support staff
- Strength of relationship with clinic staff
- Achievement of patient enrollment targets
- Strength and rigor of quality management systems
- Quality as determined by previous audit findings
The F1RST Up alliance is configured and managed to accelerate clinical trial start-up in several ways. First, redundant contracting and protracted rounds of negotiating clinical trial agreement terms and budgets are minimized or eliminated whenever possible. In 2017, F1RST Up sites averaged 25 days to approve budgets and clinical trial agreement terms compared to 41 days for non-alliance partner sites and 57 days for the comparable industry benchmark.
Second, all F1RST Up sites use central IRBs, also known as research ethics committees, that simultaneously approve multiple sites’ participation in the same protocol. Using central IRBs has a profound, positive effect on contracting timelines, site activation, and the time until enrollment of the first patient. According to CSDD, sites using a local IRB (typically academic and research centers) took an average of 390 days from site selection to reach first patient enrolled, while sites using a central IRB reached this milestone on average in about 210 days.14 In 2017, F1RST Up sites were activated in 63 days from selection on average and took another 52 days on average to reach first patient enrolled, for a total of 115 days—nearly half the time of the comparable industry benchmark of 210 days.
Third, F1RST Up is leveraged to limit the number of sites required to meet trial enrollment goals, enhance protocol design and execution, and mitigate the likelihood of later enduring amends to the protocol. After being approved, poorly designed protocols are often amended once it is realized that timelines are slipping and patient enrollment is not as projected. CSDD reported that nearly all protocols require at least one amendment, with an average of 2.3 across all development phases.15 It is also reported that each amendment includes an average of nearly seven changes to its design. Approximately 37 percent of all amendments studied by CSDD were considered avoidable. According to CSDD, “On average the direct cost to implement a single protocol amendment is approximately $500,000 in unplanned expense and adds 61 days to the project timeline. This figure undercounts the full economic impact as it does not include the cost of internal staff time dedicated to implementing each amendment, costs or fees associated with protocol language translation, and costs associated with resubmission to the local authority.”16 CSDD also estimated that the total cost for trial sponsors to implement avoidable protocol amendments in 2014 was approximately $2 billion.
F1RST Up investigators are routinely engaged to help inform protocol design. As noted by CSDD, “The primary objective in optimizing protocol design is to perform great science that can be feasibly and cost-effectively executed.”17 F1RST Up investigators work with trial sponsors to tailor clinical trial protocols to the renal patient population by starting with the patient in mind. Equally important, F1RST Up investigators work to tailor protocols to the complexities of conducting research in the dialysis delivery business and operational environments. Protocol designs that are patient-centric, site-centric, and clinic-centric are best positioned to avoid costly and time-consuming future changes and amendments.
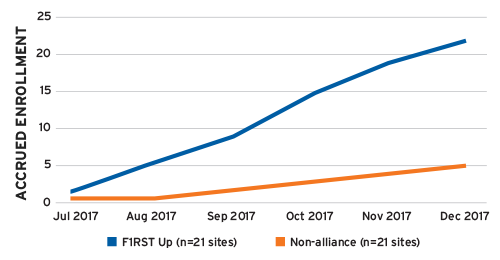
Fourth, because of their long history of conducting clinical trials, the sizable patient populations in their care, and the dramatically reduced contract and negotiation burden, F1RST Up investigators typically enroll twice as many patients into studies as non-alliance investigators. In one example, F1RST Up sites enrolled 22 patients during a six-month period compared to just five patients enrolled by an equal number of non-alliance sites during the exact same period and clinical trial (Figure 2).
F1RST Up investigators are often the highest enrolling sites among all sites globally participating in a clinical trial, including other research networks. Status as “top enroller” provides F1RST Up physicians the opportunity to develop deep subject matter expertise in a certain therapy or therapeutic area, which often leads to serving as a key opinion leader, a national clinical trial leader, and/or an expert advisor to trial sponsors. To understand best practices in clinical development, CenterWatch recently interviewed trial sponsors considered to be best in class for speed to approval. Among the factors noted for contributing to development speed was building strong relationships with investigators.18 Strong investigator-sponsor relationships typically extend beyond product approval and into product launch preparation and commercialization.
F1RST Up and the breadth of Frenova’s clinical research activities that span and accelerate the path to approving new drugs and medical devices also facilitate the commercial introduction of these new therapies into the standard of care for patients. Leveraging Frenova’s relationship-building activities across an ecosystem of renal care stakeholders—including clinical research investigators, trial sponsors, patients, patient caretakers, clinicians, and leaders in business and medicine—accelerates the speed to product launch and commercialization. This achieves Frenova’s mission to advance the science of renal therapies and help bring novel treatments to market for patients with renal impairment.
FIGURE 2 | Patient enrollment rates, F1RST Up vs. non-alliance sites
Meet Our Experts
Kurt Mussina
Vice President, Clinical Studies Operations, Frenova Renal Research
Chemist, executive, and entrepreneur, Kurt Mussina leads FMCNA’s contract research organization, a world-class network of more than 450 principal investigators across 260 research sites. A former analytical chemist and research and development scientist for Teva and Novartis pharmaceuticals, he holds a bachelor’s degree in chemistry from Montclair State University and a master of business administration from the Fuqua School of Business at Duke University.
Christina Kahn
Senior Director, Site Alliance Management, Frenova Renal Research
Christina Kahn oversees all trial activities occurring at research sites staffed by Frenova throughout the life of the trial, including oversight of research site protocol adherence, data quality, patient safety, site payment, regulatory obligations to the FDA and IRB, drug accountability, and site performance. An author of numerous publications, she is a certified clinical research professional and an affiliate member of the American Society of Nephrology. She earned her bachelor’s degree in biology with highest honors from Purdue University.
References
F1RST Up®: Accelerating Clinical Trial Start-Up
by Kurt Mussina, MBA & Christina Kahn
- Mullin, R. Tufts study finds big rise in cost of drug development. Chemical & Engineering News, November 20, 2014. https://cen.acs.org/articles/92/web/2014/11/Tufts-Study-Finds-Big-Rise.html.
- Bolten, BM. Fastest drug developers and their practices. The CenterWatch Monthly 2017 Aug 1;24(8). https://www.centerwatch.com/news-online/2017/08/01/fastest-drug-developers-practices/.
- Ibid.
- Getz K. Improving protocol design feasibility to drive drug development economics and performance. Int J Environ Res Public Health 2014 May;11(5):5069-80. doi:10.3390/ijerph110505069.
- Lamberti MJ, Chakravarthy R, Getz, KA. Assessing practices & inefficiencies with site selection, study start-up, and site activation. Applied Clinical Trials, August 5, 2016. http://www.appliedclinicaltrialsonline.com/limited-boosts-study-start.
- Lamberti MF, Wilkinson, M, Harper B., Morgan C, Getz K. Assessing study start-up practices, performance, and perceptions among sponsors and contract research organizations. Therapeutic Innovation & Regulatory Science, January 11, 2018. https://doi.org/10.1177/2168479017751403.
- Fenichel M. Site selection a continuing conundrum. The CenterWatch Monthly
2017 Sep;24(9). - Ibid.
- Lamberti et al. Assessing study start-up practices. https://doi.org/10.1177/2168479017751403.
- Bolten. Fastest drug developers. https://www.centerwatch.com/news-online/2017/08/01/fastest-drug-developers-practices/.
- Fenichel M. Site selection a continuing conundrum.
- Getz, KA. Enrollment performance: weighing the “facts.” Applied Clinical Trials 2012 May 1;21(5). http://www.appliedclinicaltrialsonline.com/enrollment-performance-weighing-facts.
- Lamberti et al. Assessing study start-up practices. https://doi.org/10.1177/2168479017751403.
- Ibid.
- Getz K. Improving protocol design feasibility. doi:10.3390/ijerph110505069.
- Ibid.
- Ibid.
- Bolten. Fastest drug developers. https://www.centerwatch.com/news-online/2017/08/01/fastest-drug-developers-practices/.



© 2006-2024 Fresenius Medical Care. All Rights Reserved.